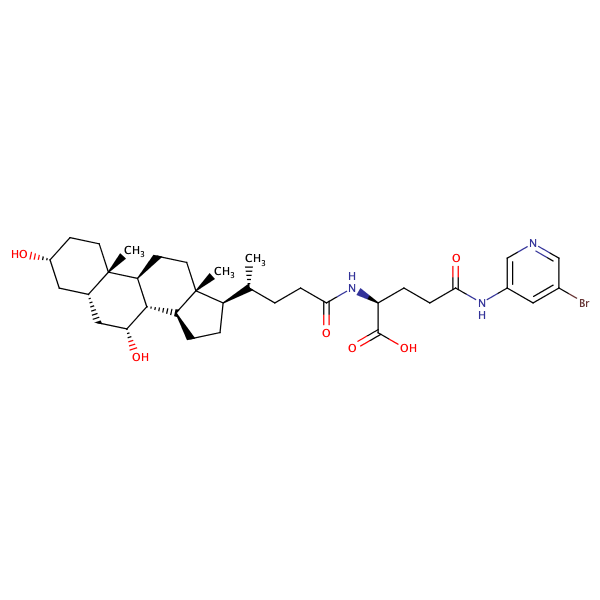
|
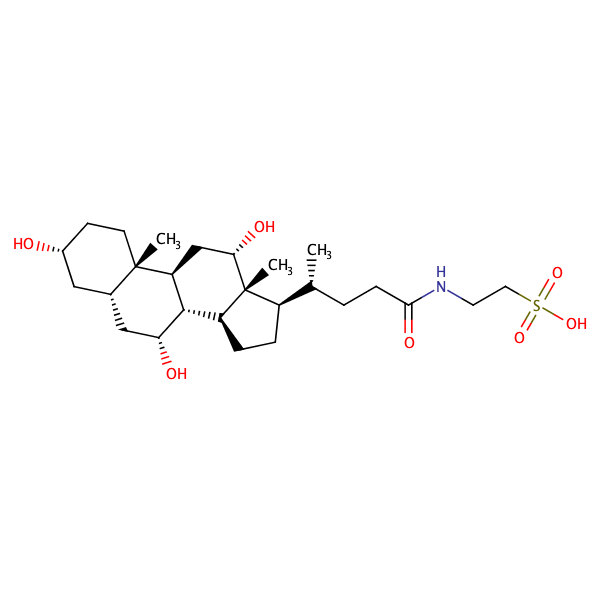
Taurocholic acid |
substrate
Km = 9.4 μM
HEK293
|
PMID: 12475240
10.1021/bi0205404
AbstractDrug intervention that prevents reabsorption of circulating bile acids by the apical (ileal) sodium/bile acid cotransporter (ASBT) may be a promising new therapy for lowering of plasma cholesterol. 2164U90 is a benzothiazepine-based competitive inhibitor of bile acid transport with K(i) values of approximately 10 and 0.068 microM for the homologous human and mouse apical transporters, respectively. Hybrid human-mouse and mouse-human transporters were engineered to identify regions involved in this 150-fold difference in the inhibition constant for 2164U90. A mouse-human chimera with only the most C-terminal hydrophobic domain and the C-terminus of the transporter originating from the human variant was found to have a sensitivity to 2164U90 inhibition similar to that of the human transporter. Conversely, a human-mouse hybrid transporter encompassing the same C-terminal region from the mouse sequence but now inserted into the human sequence demonstrated the greater inhibition seen with the mouse wild type ASBT. Amino acid substitutions, individually or in combinations, of six candidate nonconserved residues between mouse and human transporters in this C-terminal domain showed replacements of Thr294 by Ser and Val295 by Ile to be responsible for the difference in the sensitivity toward 2164U90 seen between the species. The hamster apical SBAT encompassing Ser/Ile in these positions shared the lower sensitivity to 2164U90, as seen with the human ASBT, even though it is identical to the mouse SBAT in the remaining four positions of this region. In addition, the rat ASBT which is identical to the mouse ASBT in this domain also had the high sensitivity to 2164U90 inhibition found for the mouse ASBT. Methanethiosulfonates (MTS) are known to inactivate the sodium/bile acid transporters through alkylation of a cysteine in the most C-terminal hydrophobic domain (1). Inactivation of the human ASBT due to MTS modification of cysteine 270 was shown to be largely abolished when the transporter was preincubated with 2164U90, suggesting that the binding of this benzothiazepine is in the vicinity of position 270. Thus, the domain containing the two most C-terminal putative transmembrane regions of the SBATs, H8-H9, previously shown to constitute part of the binding pocket for bile acids, interacts also with the bile acid transport competitive inhibitor, 2164U90.
|
TP-search, v2007 |
|
|
Taurocholic acid |
substrate
Uptake = 20.1 μL / mg protein
HEK293
|
PMID: 14610227
10.1124/jpet.103.060194
AbstractThe pH-sensitive activity of human organic anion transporting polypeptide OATP-B, which is expressed at the apical membrane of human small intestinal epithelial cells, was functionally characterized. When initial uptake of estrone-3-sulfate, a typical substrate of OATP, was studied kinetically, we observed an increase in V(max) with decrease of pH from 7.4 to 5.0, whereas the change in K(m) was negligible. OATP-B-mediated uptake of estrone-3-sulfate was independent of sodium, chloride, bicarbonate, or glutathione, whereas the proton ionophore carbonylcyanide p-trifluoromethoxyphenylhydrazone exhibited a pH-dependent inhibitory effect, suggesting that a proton gradient is a driving force for OATP-B. When OATP-B was expressed in human embryonic kidney 293 cells, uptake activities for anionic compounds showed various kinds of pH sensitivity. Dehydroepiandrosterone-sulfate, estrone-3-sulfate, and fexofenadine were transported by OATP-B at both neutral and acidic pH, whereas estradiol-17beta-glucuronide, acetic acid, and lactic acid were not transported at all. Transport of taurocholic acid and pravastatin by OATP-B was observed only at acidic pH, demonstrating a pH-sensitive substrate specificity of OATP-B. Because the physiological pH close to the surface of intestinal epithelial cells is acidic, the roles of OATP-B in the small intestine might be different from those in other tissues, such as liver basolateral membrane. Although the driving force for OATP-B has not been fully established, the clarification of factors, such as pH, that affect the OATP-B-activity is essential for an understanding of the physiological and pharmacological relevance of the transporter in the small intestine.
|
TP-search, v2007 |
|
|
Taurocholic acid |
substrate
Uptake = 29.0 μL / mg protein
HEK293
|
PMID: 14610227
10.1124/jpet.103.060194
AbstractThe pH-sensitive activity of human organic anion transporting polypeptide OATP-B, which is expressed at the apical membrane of human small intestinal epithelial cells, was functionally characterized. When initial uptake of estrone-3-sulfate, a typical substrate of OATP, was studied kinetically, we observed an increase in V(max) with decrease of pH from 7.4 to 5.0, whereas the change in K(m) was negligible. OATP-B-mediated uptake of estrone-3-sulfate was independent of sodium, chloride, bicarbonate, or glutathione, whereas the proton ionophore carbonylcyanide p-trifluoromethoxyphenylhydrazone exhibited a pH-dependent inhibitory effect, suggesting that a proton gradient is a driving force for OATP-B. When OATP-B was expressed in human embryonic kidney 293 cells, uptake activities for anionic compounds showed various kinds of pH sensitivity. Dehydroepiandrosterone-sulfate, estrone-3-sulfate, and fexofenadine were transported by OATP-B at both neutral and acidic pH, whereas estradiol-17beta-glucuronide, acetic acid, and lactic acid were not transported at all. Transport of taurocholic acid and pravastatin by OATP-B was observed only at acidic pH, demonstrating a pH-sensitive substrate specificity of OATP-B. Because the physiological pH close to the surface of intestinal epithelial cells is acidic, the roles of OATP-B in the small intestine might be different from those in other tissues, such as liver basolateral membrane. Although the driving force for OATP-B has not been fully established, the clarification of factors, such as pH, that affect the OATP-B-activity is essential for an understanding of the physiological and pharmacological relevance of the transporter in the small intestine.
|
TP-search, v2007 |
|
|
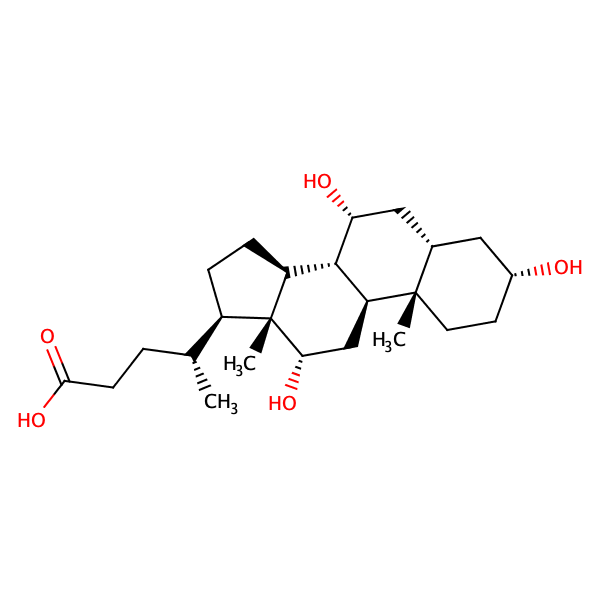
Taurocholic acid |
substrate
Km(app) = 13.3 μM
COS
|
PMID: 9458785
http://ajpgi.physiology.org/content/274/1/G157.long
AbstractThe enterohepatic circulation of bile acids is maintained by Na+-dependent transport mechanisms. To better understand these processes, a full-length human ileal Na+-bile acid cotransporter cDNA was identified using rapid amplification of cDNA ends and genomic cloning techniques. Using Northern blot analysis to determine its tissue expression, we readily detected the ileal Na+-bile acid cotransporter mRNA in terminal ileum and kidney. Direct cloning and mapping of the transcriptional start sites confirmed that the kidney cDNA was identical to the ileal Na+-bile acid cotransporter. In transiently transfected COS cells, ileal Na+-bile acid cotransporter-mediated taurocholate uptake was strictly Na+ dependent and chloride independent. Analysis of the substrate specificity in transfected COS or CHO cells showed that both conjugated and unconjugated bile acids are efficiently transported. When the inhibition constants for other potential substrates such as estrone-3-sulfate were determined, the ileal Na+-bile acid cotransporter exhibited a narrower substrate specificity than the related liver Na+-bile acid cotransporter. Whereas the multispecific liver Na+-bile acid cotransporter may participate in hepatic clearance of organic anion metabolites and xenobiotics, the ileal and renal Na+-bile acid cotransporter retains a narrow specificity for reclamation of bile acids.
|
TP-search, v2007 |
|
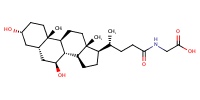
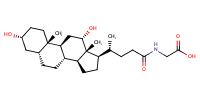
Glycoursodeoxycholate |
substrate
Km(app) = 4.1 μM
COS
|
PMID: 9458785
http://ajpgi.physiology.org/content/274/1/G157.long
AbstractThe enterohepatic circulation of bile acids is maintained by Na+-dependent transport mechanisms. To better understand these processes, a full-length human ileal Na+-bile acid cotransporter cDNA was identified using rapid amplification of cDNA ends and genomic cloning techniques. Using Northern blot analysis to determine its tissue expression, we readily detected the ileal Na+-bile acid cotransporter mRNA in terminal ileum and kidney. Direct cloning and mapping of the transcriptional start sites confirmed that the kidney cDNA was identical to the ileal Na+-bile acid cotransporter. In transiently transfected COS cells, ileal Na+-bile acid cotransporter-mediated taurocholate uptake was strictly Na+ dependent and chloride independent. Analysis of the substrate specificity in transfected COS or CHO cells showed that both conjugated and unconjugated bile acids are efficiently transported. When the inhibition constants for other potential substrates such as estrone-3-sulfate were determined, the ileal Na+-bile acid cotransporter exhibited a narrower substrate specificity than the related liver Na+-bile acid cotransporter. Whereas the multispecific liver Na+-bile acid cotransporter may participate in hepatic clearance of organic anion metabolites and xenobiotics, the ileal and renal Na+-bile acid cotransporter retains a narrow specificity for reclamation of bile acids.
|
TP-search, v2007 |
|
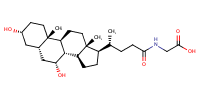
Glycodeoxycholate |
substrate
Km(app) = 2.0 μM
COS
|
PMID: 9458785
http://ajpgi.physiology.org/content/274/1/G157.long
AbstractThe enterohepatic circulation of bile acids is maintained by Na+-dependent transport mechanisms. To better understand these processes, a full-length human ileal Na+-bile acid cotransporter cDNA was identified using rapid amplification of cDNA ends and genomic cloning techniques. Using Northern blot analysis to determine its tissue expression, we readily detected the ileal Na+-bile acid cotransporter mRNA in terminal ileum and kidney. Direct cloning and mapping of the transcriptional start sites confirmed that the kidney cDNA was identical to the ileal Na+-bile acid cotransporter. In transiently transfected COS cells, ileal Na+-bile acid cotransporter-mediated taurocholate uptake was strictly Na+ dependent and chloride independent. Analysis of the substrate specificity in transfected COS or CHO cells showed that both conjugated and unconjugated bile acids are efficiently transported. When the inhibition constants for other potential substrates such as estrone-3-sulfate were determined, the ileal Na+-bile acid cotransporter exhibited a narrower substrate specificity than the related liver Na+-bile acid cotransporter. Whereas the multispecific liver Na+-bile acid cotransporter may participate in hepatic clearance of organic anion metabolites and xenobiotics, the ileal and renal Na+-bile acid cotransporter retains a narrow specificity for reclamation of bile acids.
|
TP-search, v2007 |
|
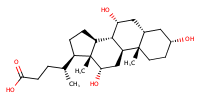
Glycochenodeoxycholic acid |
substrate
Km(app) = 5.7 μM
COS
|
PMID: 9458785
http://ajpgi.physiology.org/content/274/1/G157.long
AbstractThe enterohepatic circulation of bile acids is maintained by Na+-dependent transport mechanisms. To better understand these processes, a full-length human ileal Na+-bile acid cotransporter cDNA was identified using rapid amplification of cDNA ends and genomic cloning techniques. Using Northern blot analysis to determine its tissue expression, we readily detected the ileal Na+-bile acid cotransporter mRNA in terminal ileum and kidney. Direct cloning and mapping of the transcriptional start sites confirmed that the kidney cDNA was identical to the ileal Na+-bile acid cotransporter. In transiently transfected COS cells, ileal Na+-bile acid cotransporter-mediated taurocholate uptake was strictly Na+ dependent and chloride independent. Analysis of the substrate specificity in transfected COS or CHO cells showed that both conjugated and unconjugated bile acids are efficiently transported. When the inhibition constants for other potential substrates such as estrone-3-sulfate were determined, the ileal Na+-bile acid cotransporter exhibited a narrower substrate specificity than the related liver Na+-bile acid cotransporter. Whereas the multispecific liver Na+-bile acid cotransporter may participate in hepatic clearance of organic anion metabolites and xenobiotics, the ileal and renal Na+-bile acid cotransporter retains a narrow specificity for reclamation of bile acids.
|
TP-search, v2007 |
|
Cholic acid |
substrate
Km(app) = 33.3 μM
COS
|
PMID: 9458785
http://ajpgi.physiology.org/content/274/1/G157.long
AbstractThe enterohepatic circulation of bile acids is maintained by Na+-dependent transport mechanisms. To better understand these processes, a full-length human ileal Na+-bile acid cotransporter cDNA was identified using rapid amplification of cDNA ends and genomic cloning techniques. Using Northern blot analysis to determine its tissue expression, we readily detected the ileal Na+-bile acid cotransporter mRNA in terminal ileum and kidney. Direct cloning and mapping of the transcriptional start sites confirmed that the kidney cDNA was identical to the ileal Na+-bile acid cotransporter. In transiently transfected COS cells, ileal Na+-bile acid cotransporter-mediated taurocholate uptake was strictly Na+ dependent and chloride independent. Analysis of the substrate specificity in transfected COS or CHO cells showed that both conjugated and unconjugated bile acids are efficiently transported. When the inhibition constants for other potential substrates such as estrone-3-sulfate were determined, the ileal Na+-bile acid cotransporter exhibited a narrower substrate specificity than the related liver Na+-bile acid cotransporter. Whereas the multispecific liver Na+-bile acid cotransporter may participate in hepatic clearance of organic anion metabolites and xenobiotics, the ileal and renal Na+-bile acid cotransporter retains a narrow specificity for reclamation of bile acids.
|
TP-search, v2007 |
|
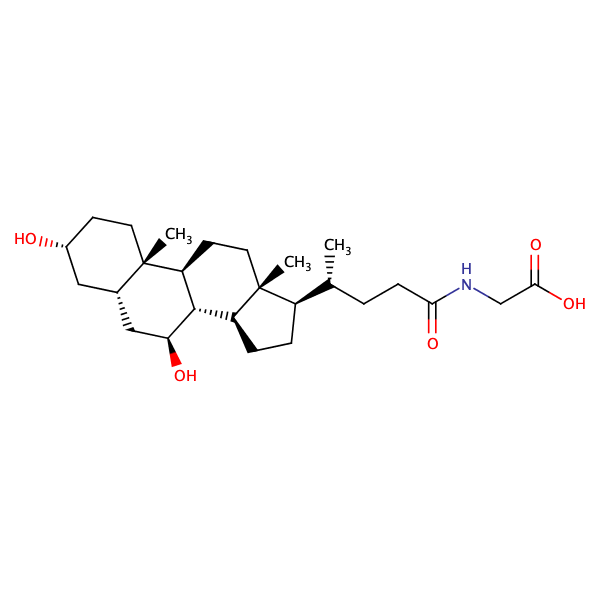
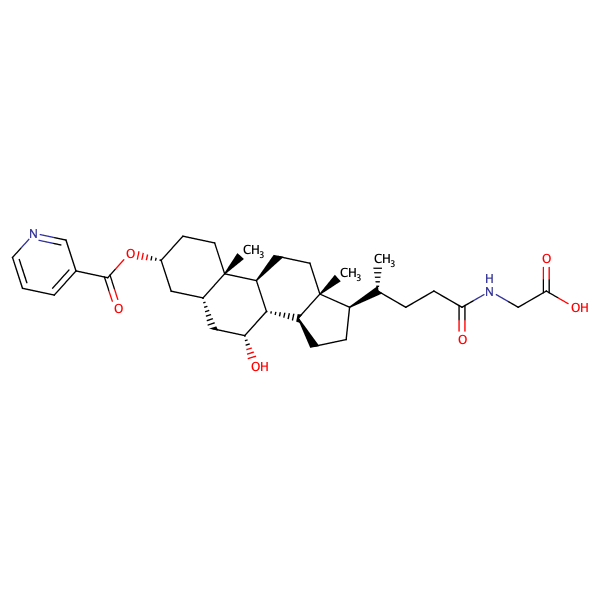
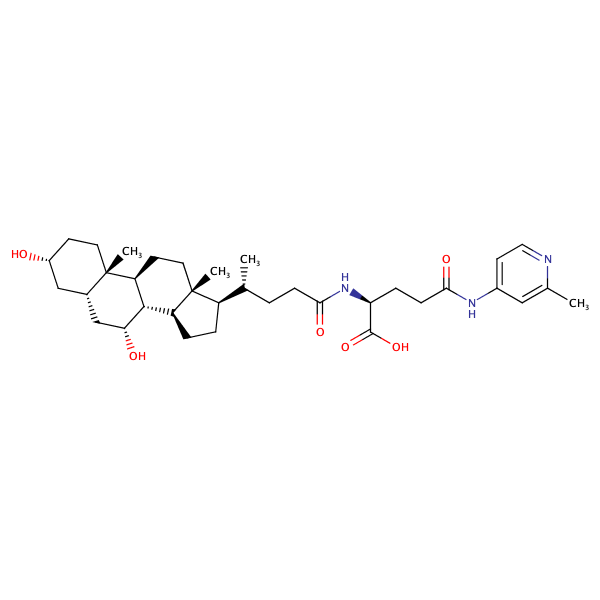
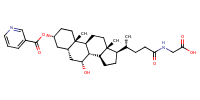
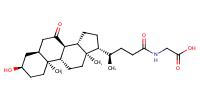
Glycine, N-[(3alpha,5beta,7alpha)-7-hydroxy-24-oxo-3-[(3-pyridinylcarbonyl)oxy]cholan-24-yl]- |
substrate
Km = 8.49 μM
MDCK
|
PMID: 22387310
10.1016/j.ejps.2012.02.012
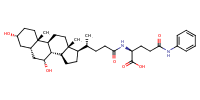
AbstractThe apical sodium dependent bile acid transporter (ASBT) and sodium-taurocholate cotransporting polypeptide (NTCP) are potential prodrug targets, but the structural requirements for these transporters are incompletely defined. The objective of this study was to evaluate the effect of C-3 and C-7 substitution on bile acid interaction with these bile acid transporters. Nineteen bile acid analogs were tested against ASBT and NTCP for binding, as well as translocation. Results indicated that ASBT and NTCP accommodated a wide range of substituents for binding, but all major C-7 modifications resulted in analogs that did not demonstrate active uptake by either ASBT or NTCP. A C-3 modification that was not tolerated at C-7 still afforded translocation via ASBT and NTCP, confirming the relative unacceptability of C-7 modification. Both ASBT and NTCP demonstrated a generally similar binding potency. Results suggest that drug conjugation to the C-3 hydroxyl group, rather than C-7, has potential to lead to a successful prodrug targeting ASBT and NTCP.
|
literature |
|
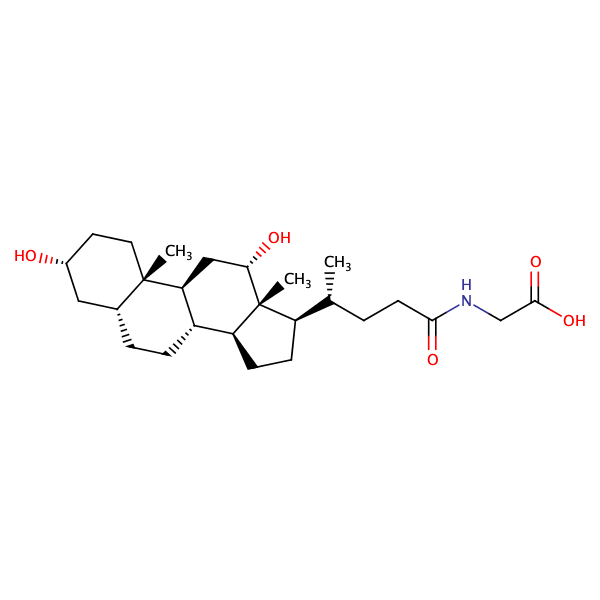
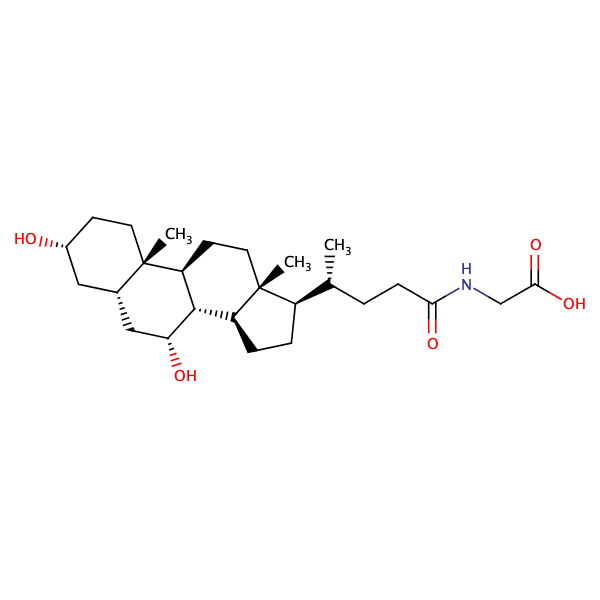
Glycine, N-[(3alpha,5beta)-3-hydroxy-7,24-dioxocholan-24-yl]- |
substrate
Km = 34.0 μM
MDCK
|
PMID: 22387310
10.1016/j.ejps.2012.02.012
AbstractThe apical sodium dependent bile acid transporter (ASBT) and sodium-taurocholate cotransporting polypeptide (NTCP) are potential prodrug targets, but the structural requirements for these transporters are incompletely defined. The objective of this study was to evaluate the effect of C-3 and C-7 substitution on bile acid interaction with these bile acid transporters. Nineteen bile acid analogs were tested against ASBT and NTCP for binding, as well as translocation. Results indicated that ASBT and NTCP accommodated a wide range of substituents for binding, but all major C-7 modifications resulted in analogs that did not demonstrate active uptake by either ASBT or NTCP. A C-3 modification that was not tolerated at C-7 still afforded translocation via ASBT and NTCP, confirming the relative unacceptability of C-7 modification. Both ASBT and NTCP demonstrated a generally similar binding potency. Results suggest that drug conjugation to the C-3 hydroxyl group, rather than C-7, has potential to lead to a successful prodrug targeting ASBT and NTCP.
|
literature |
|
|
Glycine, N-[(3alpha,5beta,7alpha)-7-hydroxy-24-oxo-3-[(3-pyridinylcarbonyl)oxy]cholan-24-yl]- |
substrate
Vmax = 0.000242 nmol s-1 cm-2
MDCK
|
PMID: 22387310
10.1016/j.ejps.2012.02.012
AbstractThe apical sodium dependent bile acid transporter (ASBT) and sodium-taurocholate cotransporting polypeptide (NTCP) are potential prodrug targets, but the structural requirements for these transporters are incompletely defined. The objective of this study was to evaluate the effect of C-3 and C-7 substitution on bile acid interaction with these bile acid transporters. Nineteen bile acid analogs were tested against ASBT and NTCP for binding, as well as translocation. Results indicated that ASBT and NTCP accommodated a wide range of substituents for binding, but all major C-7 modifications resulted in analogs that did not demonstrate active uptake by either ASBT or NTCP. A C-3 modification that was not tolerated at C-7 still afforded translocation via ASBT and NTCP, confirming the relative unacceptability of C-7 modification. Both ASBT and NTCP demonstrated a generally similar binding potency. Results suggest that drug conjugation to the C-3 hydroxyl group, rather than C-7, has potential to lead to a successful prodrug targeting ASBT and NTCP.
|
literature |
|
|
Glycine, N-[(3alpha,5beta)-3-hydroxy-7,24-dioxocholan-24-yl]- |
substrate
Vmax = 9.1e-05 nmol s-1 cm-2
MDCK
|
PMID: 22387310
10.1016/j.ejps.2012.02.012
AbstractThe apical sodium dependent bile acid transporter (ASBT) and sodium-taurocholate cotransporting polypeptide (NTCP) are potential prodrug targets, but the structural requirements for these transporters are incompletely defined. The objective of this study was to evaluate the effect of C-3 and C-7 substitution on bile acid interaction with these bile acid transporters. Nineteen bile acid analogs were tested against ASBT and NTCP for binding, as well as translocation. Results indicated that ASBT and NTCP accommodated a wide range of substituents for binding, but all major C-7 modifications resulted in analogs that did not demonstrate active uptake by either ASBT or NTCP. A C-3 modification that was not tolerated at C-7 still afforded translocation via ASBT and NTCP, confirming the relative unacceptability of C-7 modification. Both ASBT and NTCP demonstrated a generally similar binding potency. Results suggest that drug conjugation to the C-3 hydroxyl group, rather than C-7, has potential to lead to a successful prodrug targeting ASBT and NTCP.
|
literature |
|
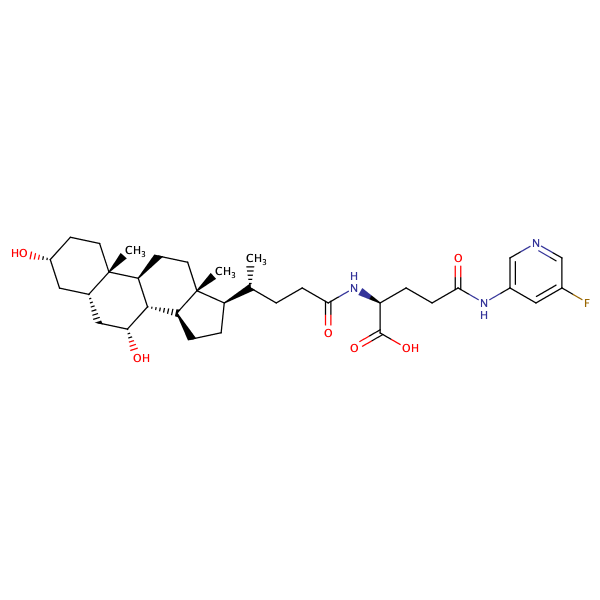
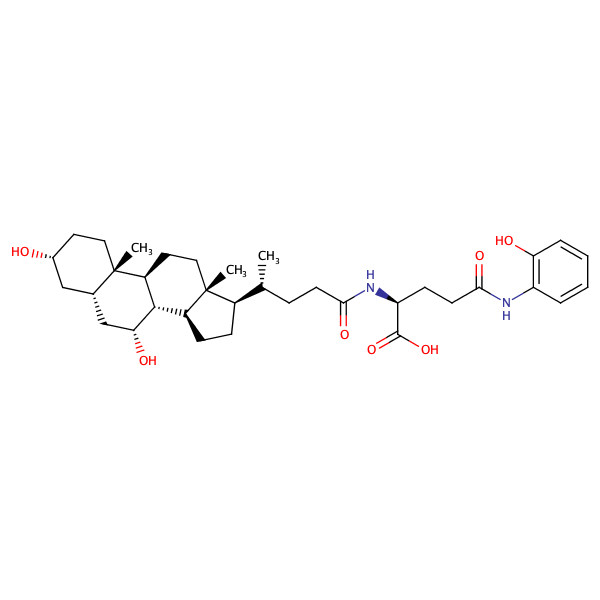
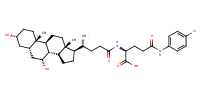
L-Glutamine, N2-[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]-N5-phenyl- |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
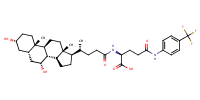
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
|
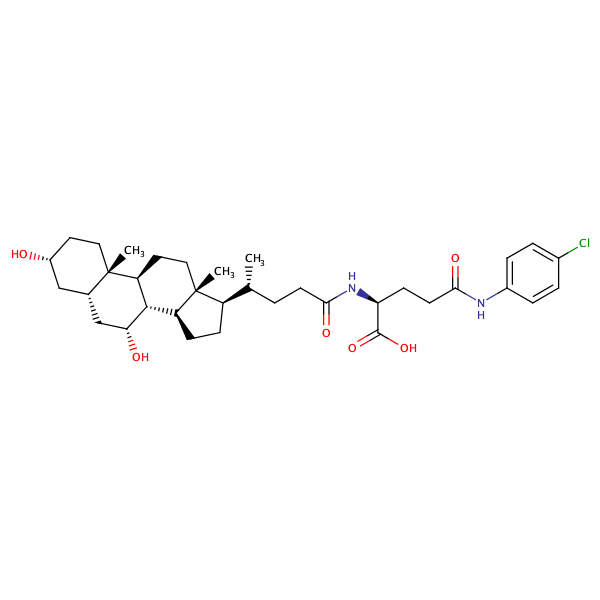
L-Glutamine, N5-(4-chlorophenyl)-N2-[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]- |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
|
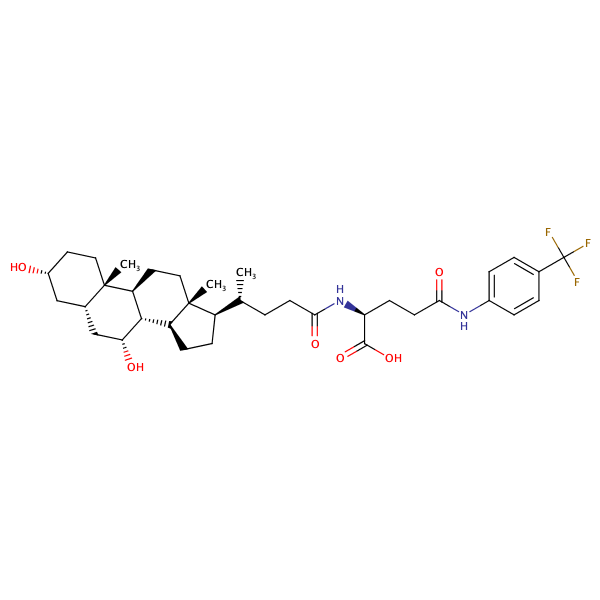
L-Glutamine, N2-[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]-N5-[4-(trifluoromethyl)phenyl]- |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
|
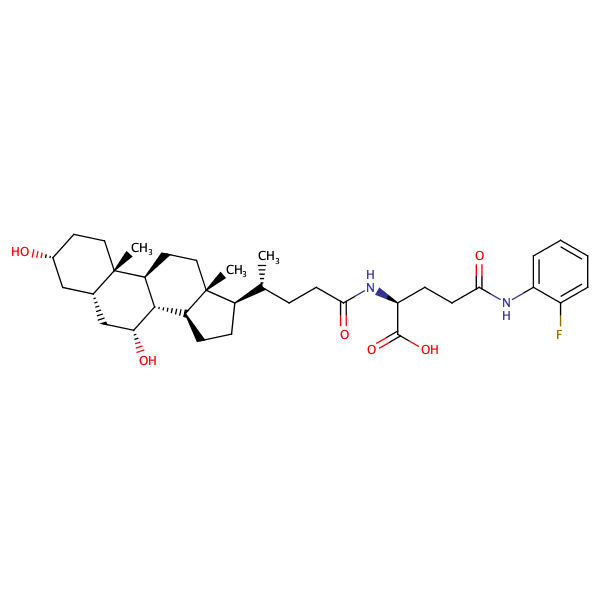
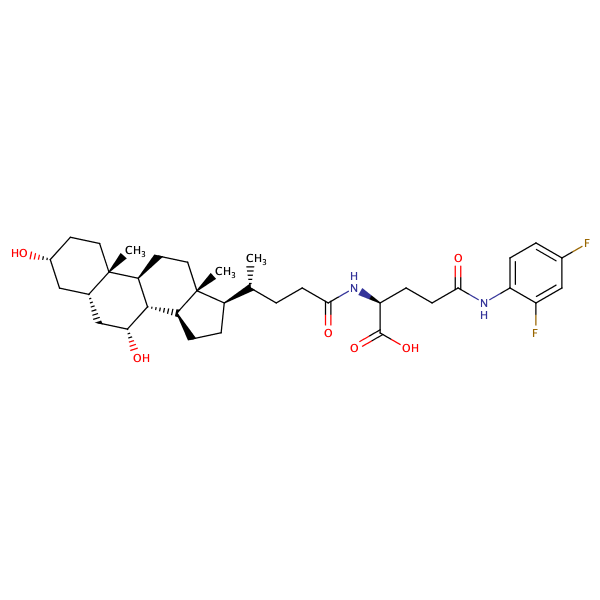
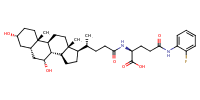
L-Glutamine, N2-[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]-N5-(2-fluorophenyl)- |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
|
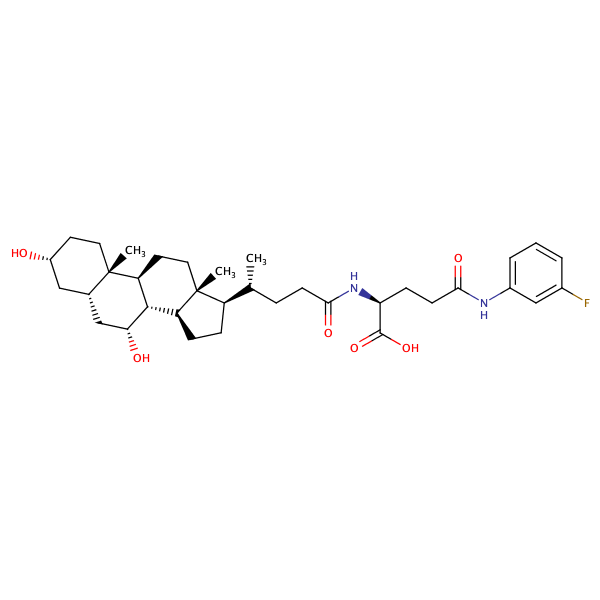
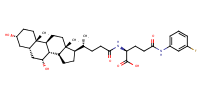
L-Glutamine, N2-[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]-N5-(3-fluorophenyl)- |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
|
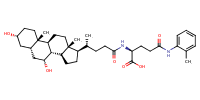
L-Glutamine, N2-[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]-N5-(4-fluorophenyl)- |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
|
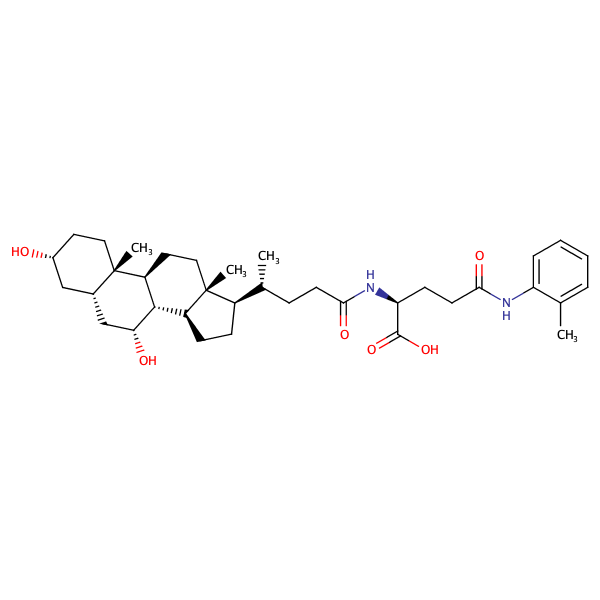
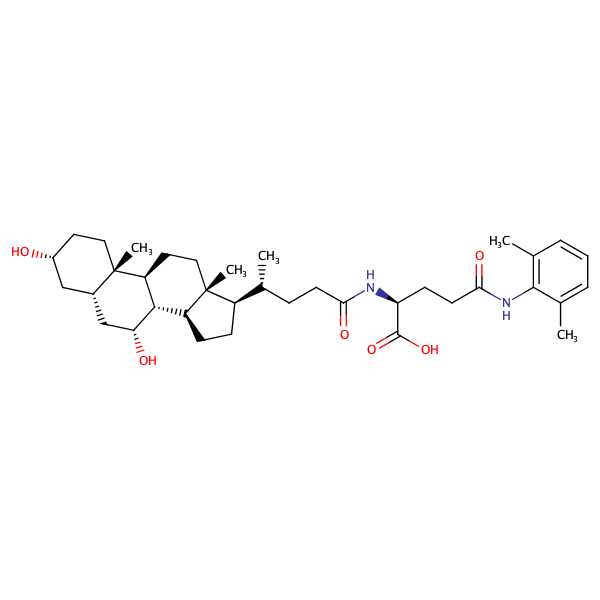
L-Glutamine, N2-[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]-N5-(2-methylphenyl)- |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |

|
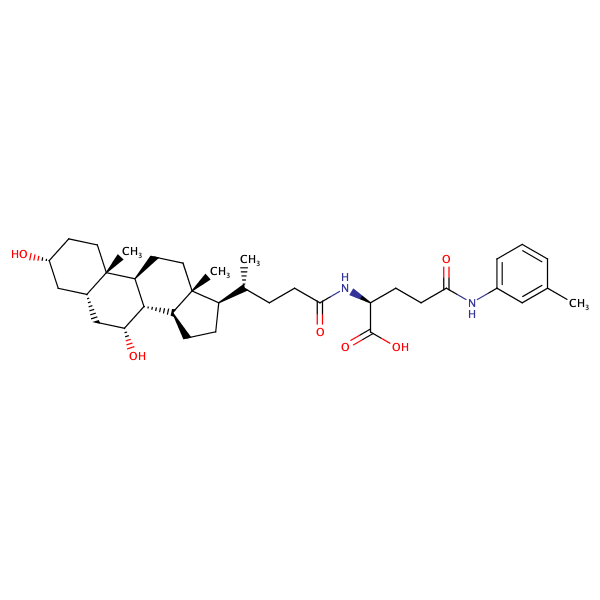
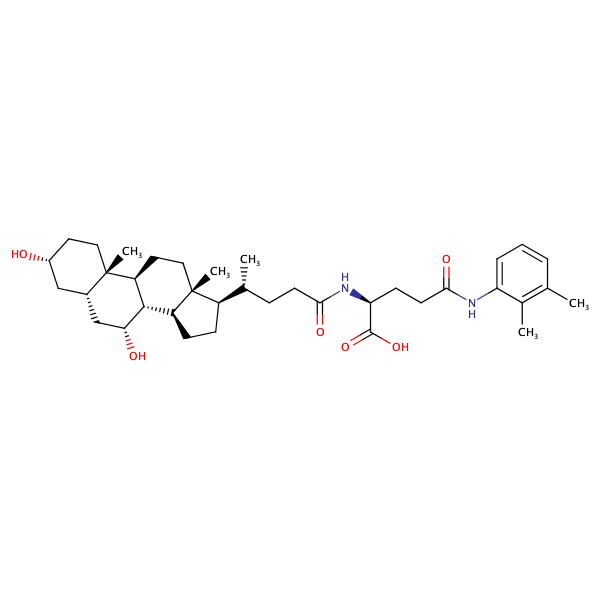
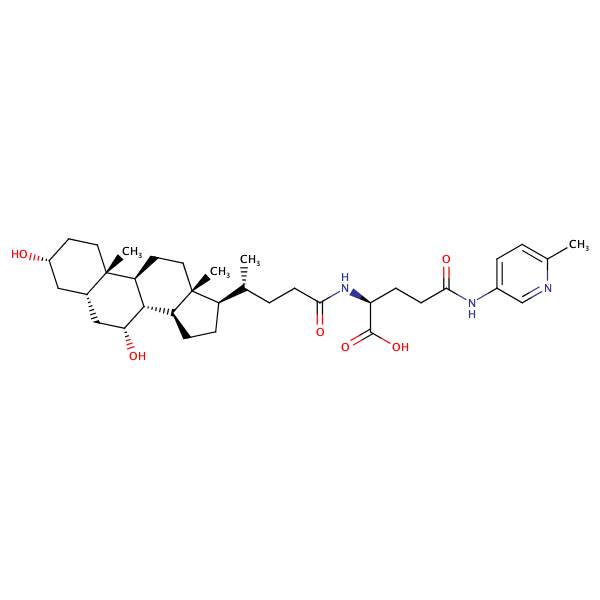
L-Glutamine, N2-[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]-N5-(3-methylphenyl)- |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |

|
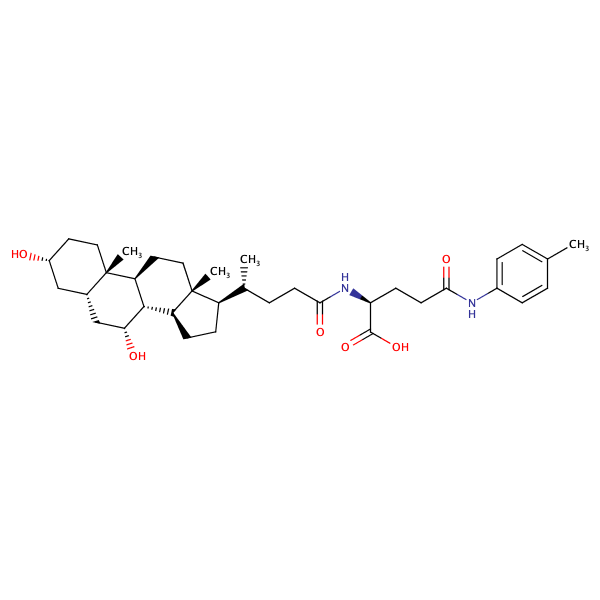
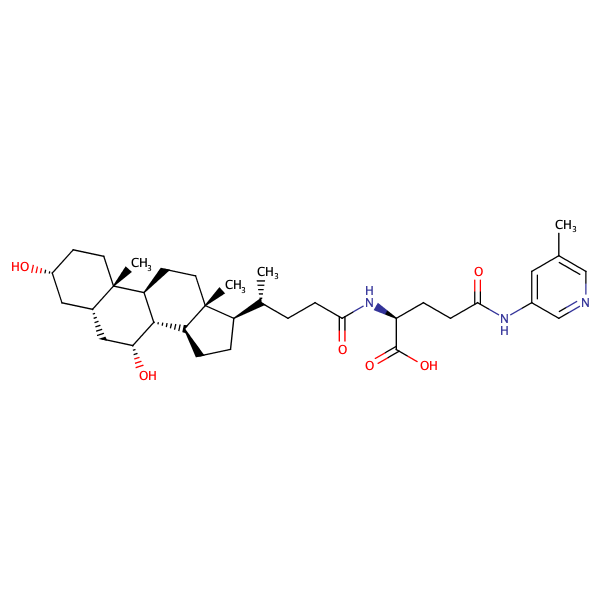
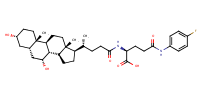
L-Glutamine, N2-[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]-N5-(4-methylphenyl)- |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
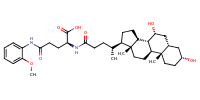
|
L-Glutamine, N2-[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]-N5-(2-methoxyphenyl)- |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
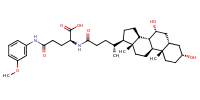
|
L-Glutamine, N2-[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]-N5-(3-methoxyphenyl)- |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
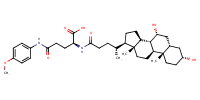
|
L-Glutamine, N2-[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]-N5-(4-methoxyphenyl)- |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
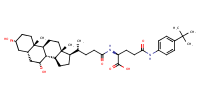
|
L-Glutamine, N2-[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]-N5-[4-(1,1-dimethylethyl)phenyl]- |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
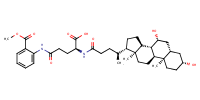
|
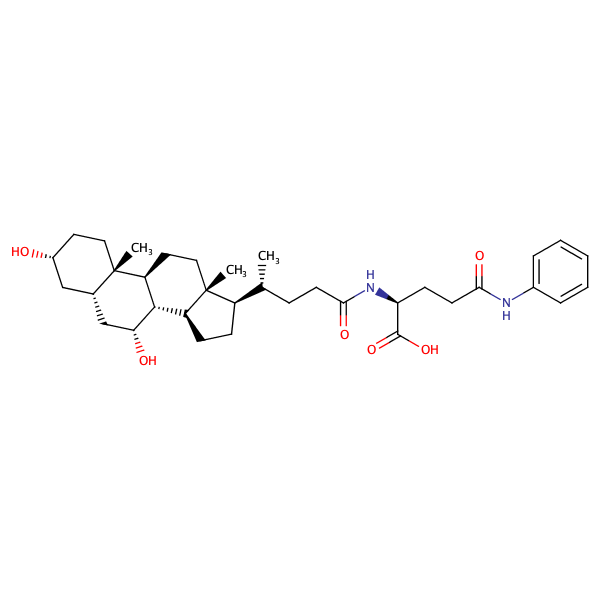
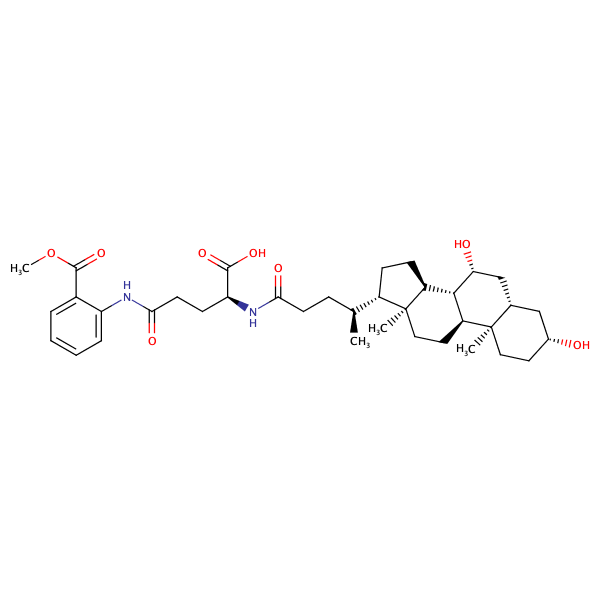
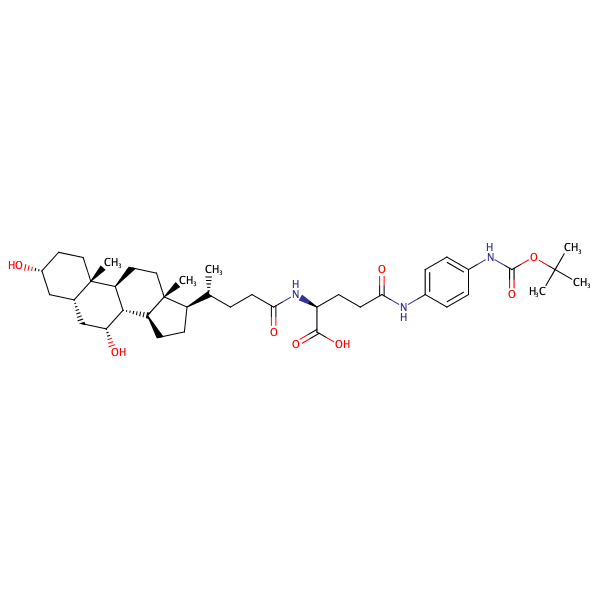
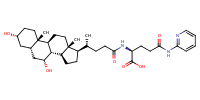
Benzoic acid, 2-[[(4S)-4-carboxy-4-[[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]amino]-1-oxobutyl]amino]-, 1-methyl ester |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
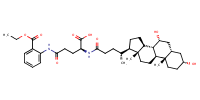
|
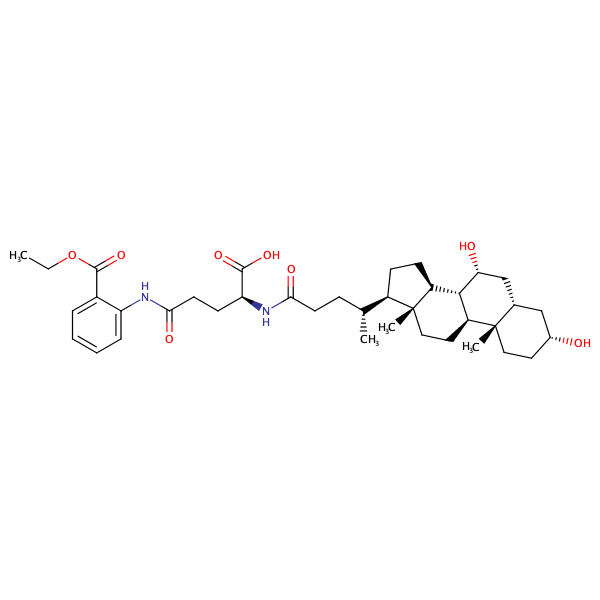
Benzoic acid, 2-[[(4S)-4-carboxy-4-[[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]amino]-1-oxobutyl]amino]-, 1-ethyl ester |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
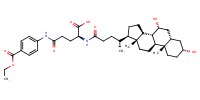
|
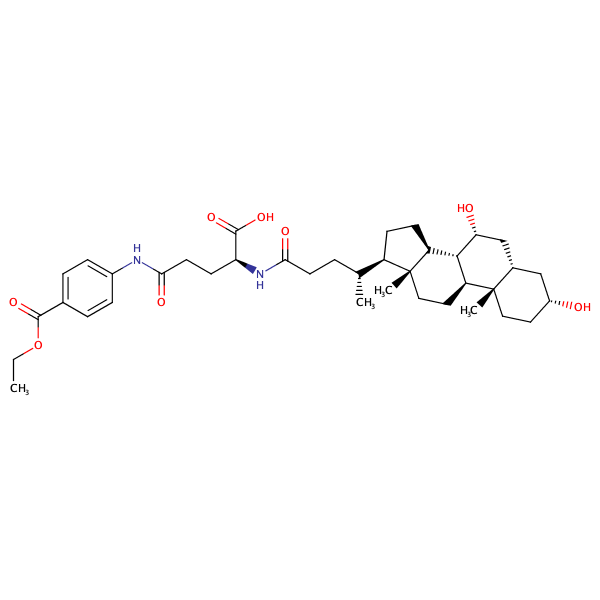
Benzoic acid, 4-[[(4S)-4-carboxy-4-[[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]amino]-1-oxobutyl]amino]-, 1-ethyl ester |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
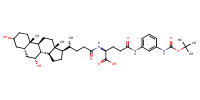
|
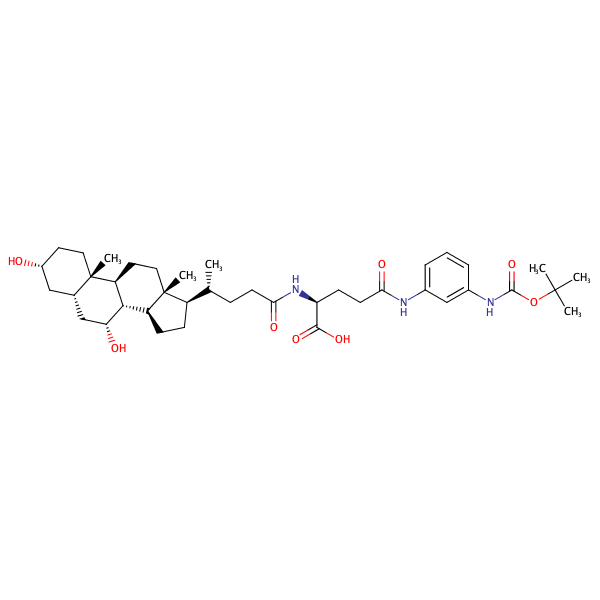
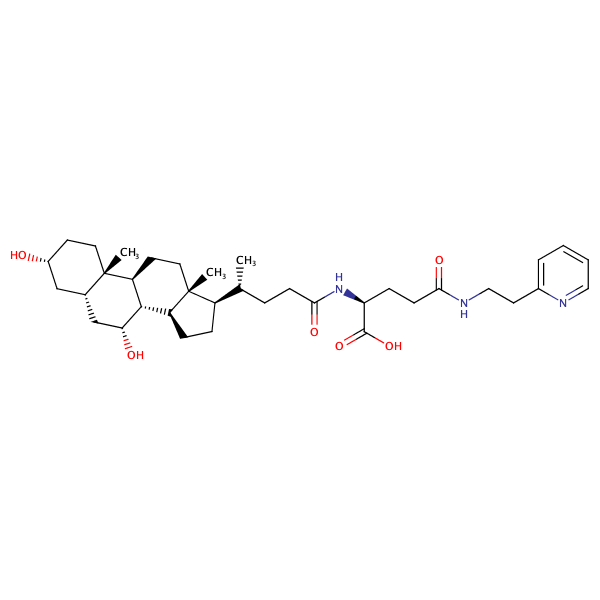
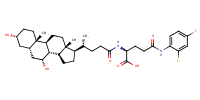
L-Glutamine, N2-[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]-N5-[3-[[(1,1-dimethylethoxy)carbonyl]amino]phenyl]- |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
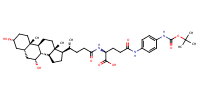
|
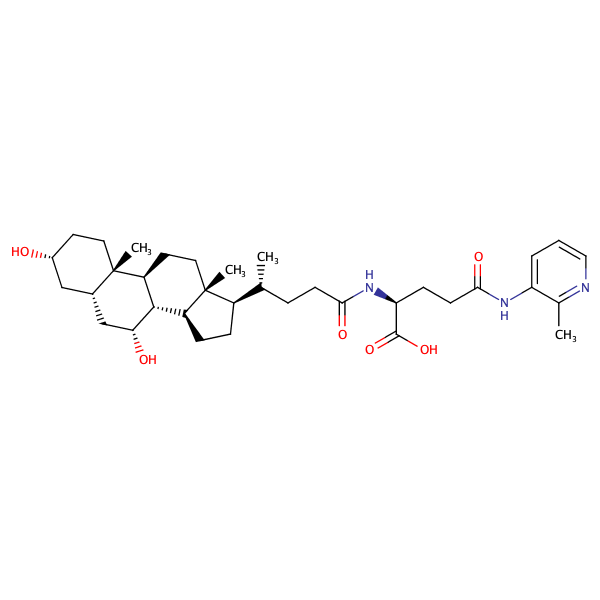
L-Glutamine, N2-[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]-N5-[4-[[(1,1-dimethylethoxy)carbonyl]amino]phenyl]- |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
|
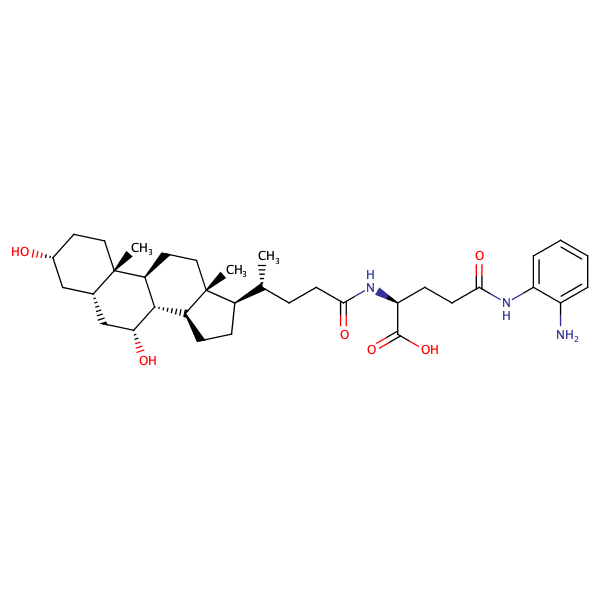
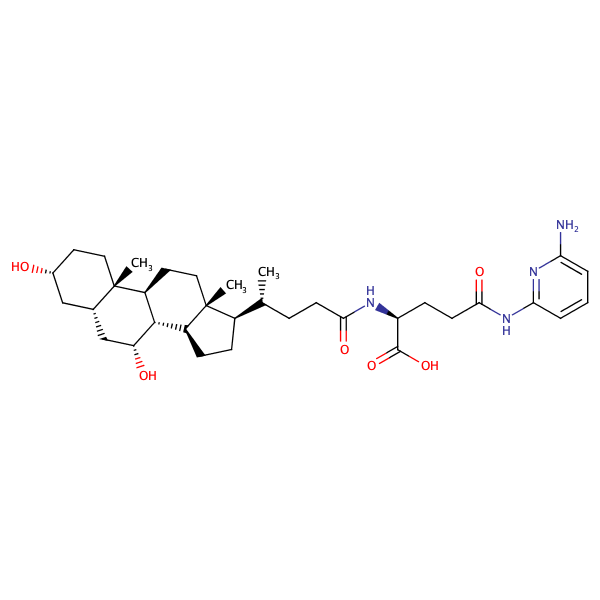
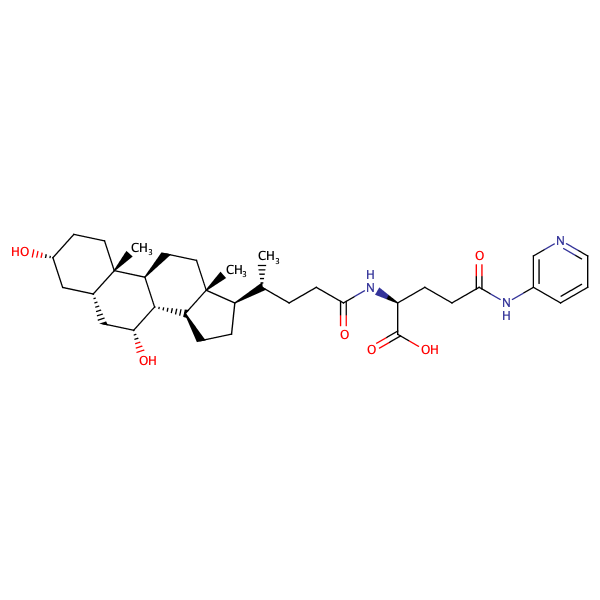
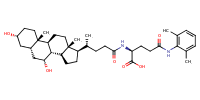
L-Glutamine, N5-(2-aminophenyl)-N2-[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]- |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |

|
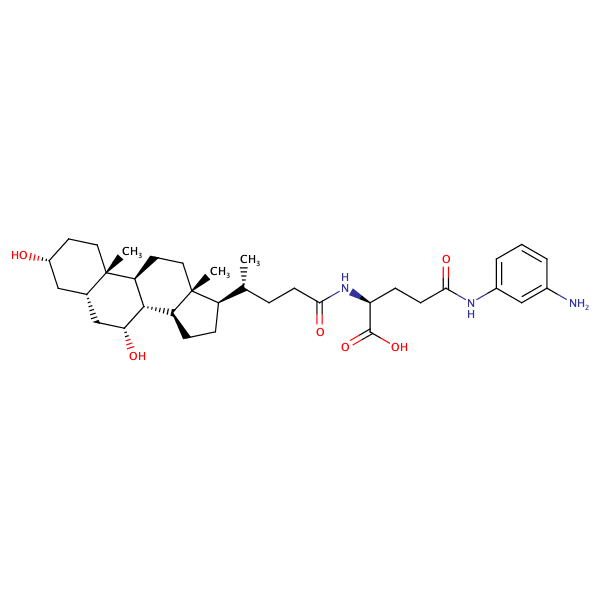
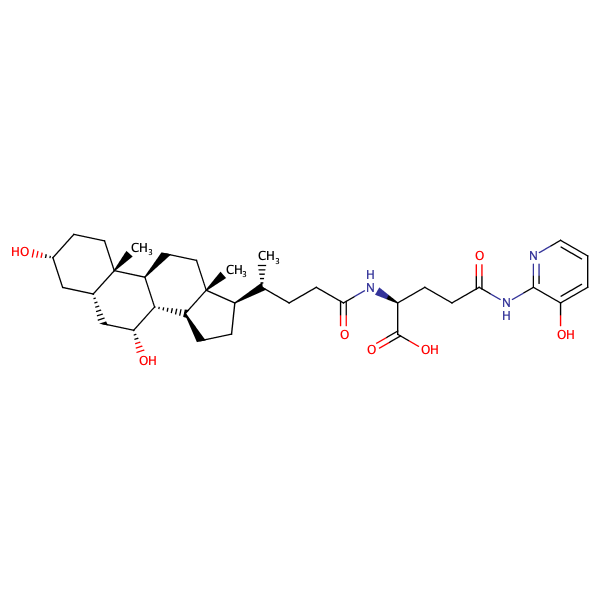
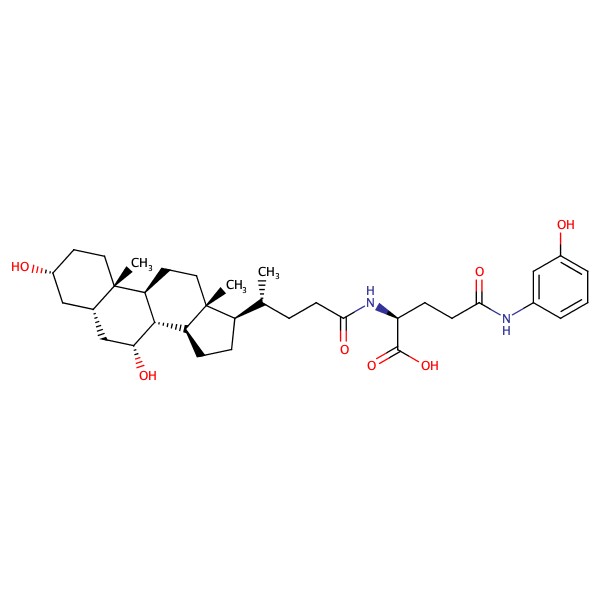
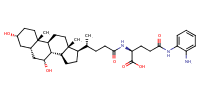
L-Glutamine, N5-(3-aminophenyl)-N2-[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]- |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |

|
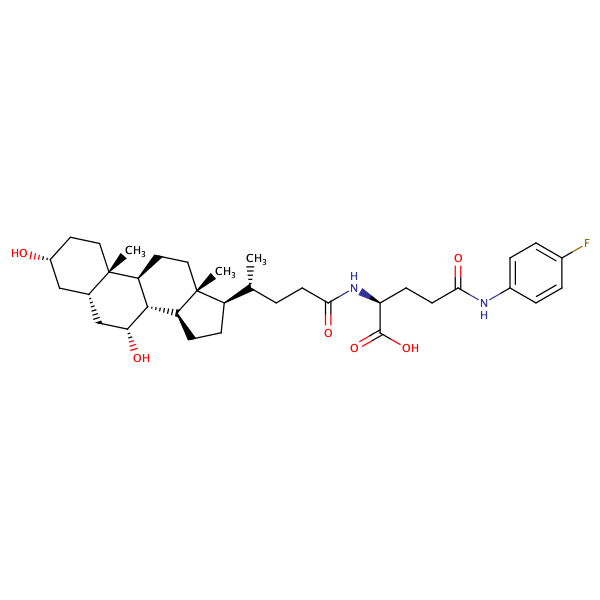
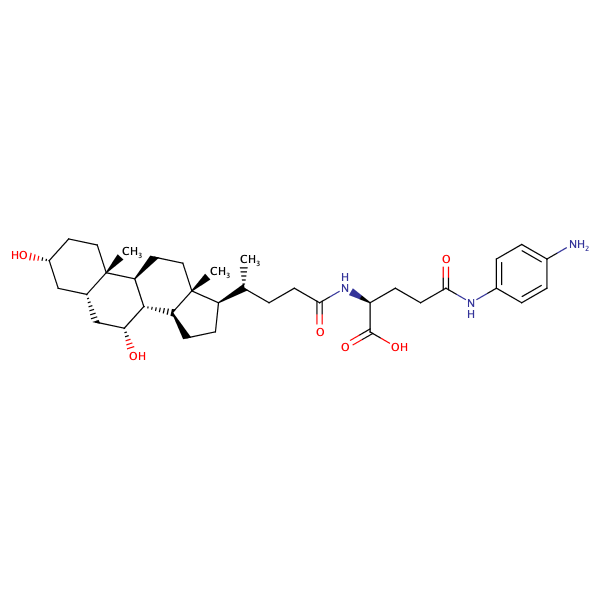
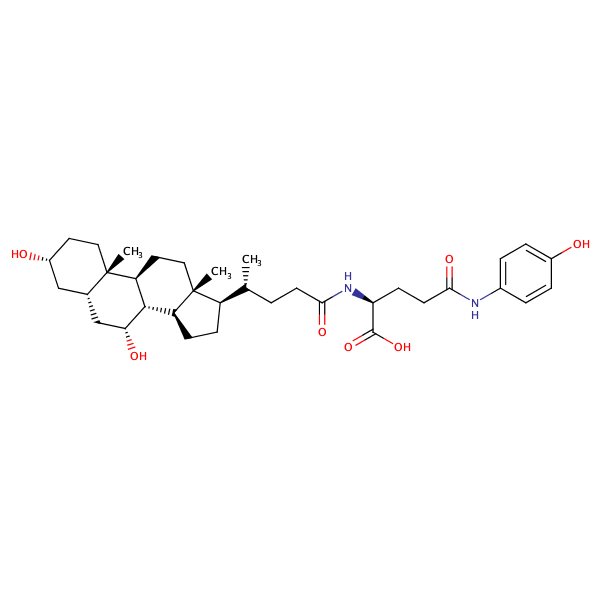
L-Glutamine, N5-(4-aminophenyl)-N2-[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]- |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
|
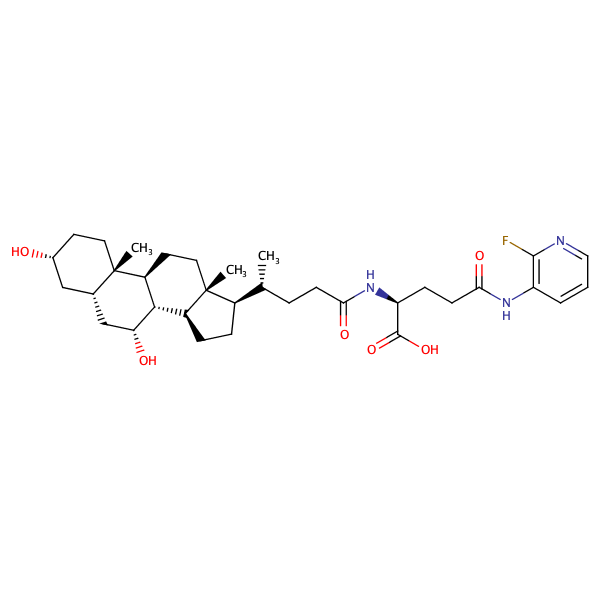
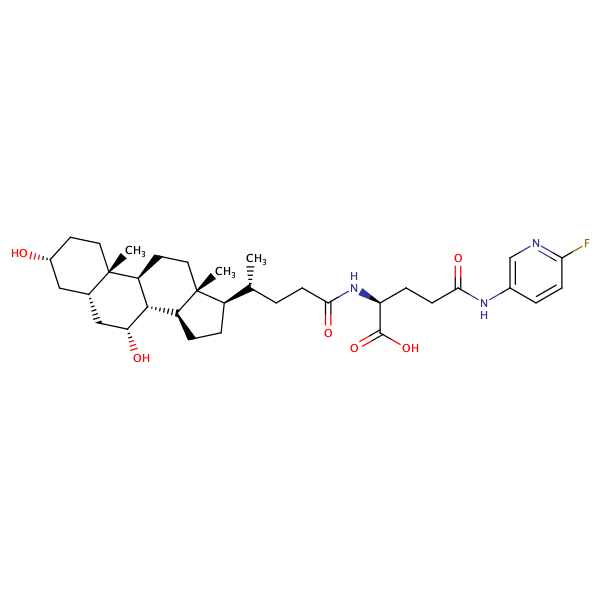
L-Glutamine, N5-(2,4-difluorophenyl)-N2-[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]- |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
|
L-Glutamine, N2-[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]-N5-(2,6-dimethylphenyl)- |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |

|
L-Glutamine, N2-[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]-N5-(2,3-dimethylphenyl)- |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
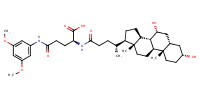
|
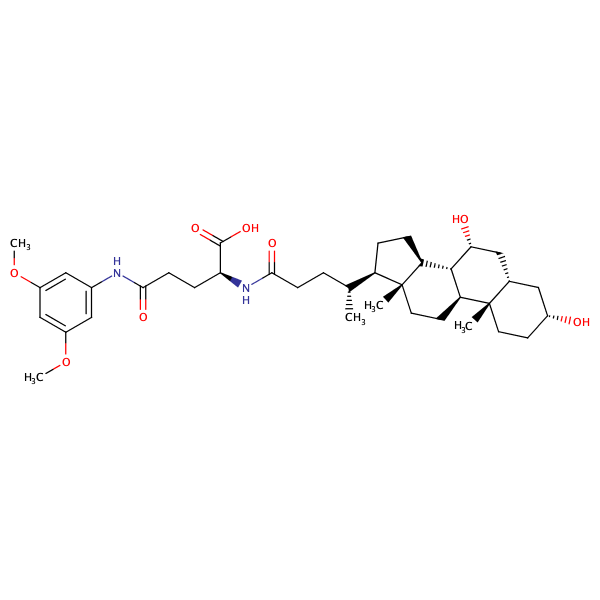
L-Glutamine, N2-[(3alpha,5beta,7alpha)-3,7-dihydroxy-24-oxocholan-24-yl]-N5-(3,5-dimethoxyphenyl)- |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
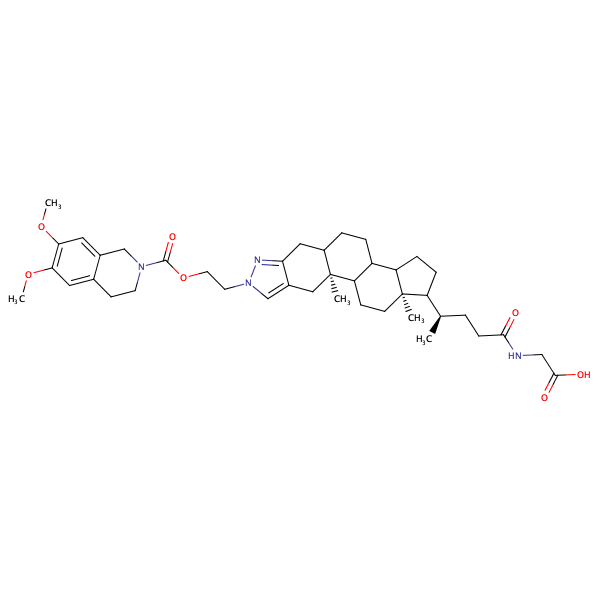
![Glycine,N-[(5-beta)-1'-[2-[[(3,4-dihydro-6,7-dimethoxy-2(1H)-isoquinolinyl)carbonyl]oxy]ethyl]-24-oxo-1'H-cholano[3,2-c]pyrazol-24-yl]-](/static/metrabaseui/img/cmpds/h100w200/mbcd0003472.png)
|
Glycine,N-[(5-beta)-1'-[2-[[(3,4-dihydro-6,7-dimethoxy-2(1H)-isoquinolinyl)carbonyl]oxy]ethyl]-24-oxo-1'H-cholano[3,2-c]pyrazol-24-yl]- |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
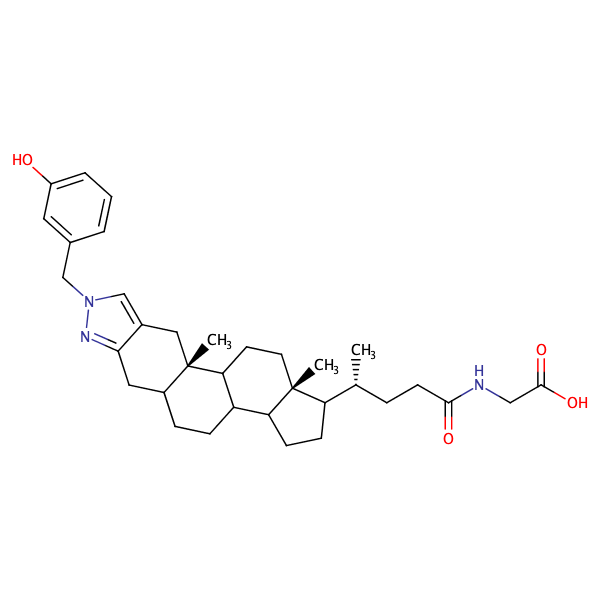
![Glycine,N-[(5-beta)-1'-[(3-hydroxyphenyl)methyl]-24-oxo-1'H-cholano[3,2-c]pyrazol-24-yl]-](/static/metrabaseui/img/cmpds/h100w200/mbcd0003404.png)
|
Glycine,N-[(5-beta)-1'-[(3-hydroxyphenyl)methyl]-24-oxo-1'H-cholano[3,2-c]pyrazol-24-yl]- |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
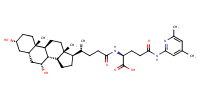
|
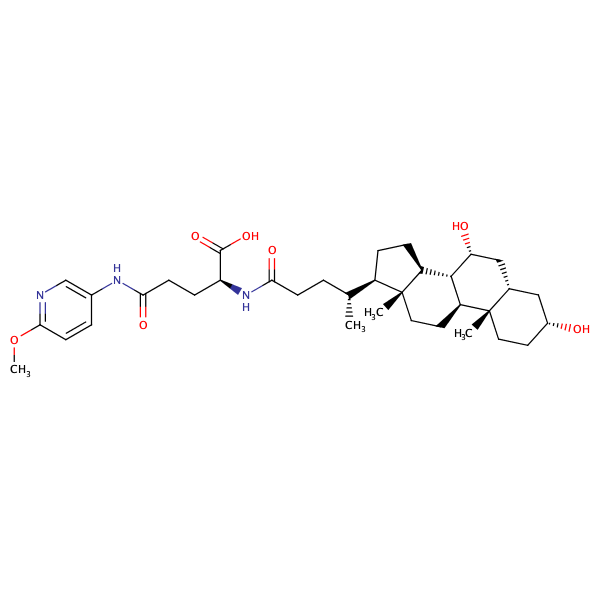
glu-CDCA-aminopyridine_conj1 |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
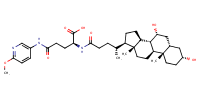
|
glu-CDCA-aminopyridine_conj10 |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
|
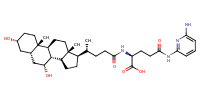
glu-CDCA-aminopyridine_conj14 |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
|
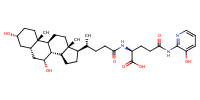
glu-CDCA-aminopyridine_conj15 |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
|
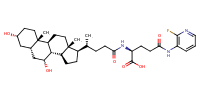
glu-CDCA-aminopyridine_conj16 |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
|
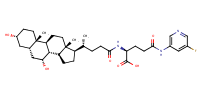
glu-CDCA-aminopyridine_conj18 |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
|
glu-CDCA-aminopyridine_conj19 |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
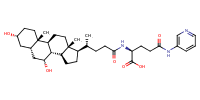
|
glu-CDCA-aminopyridine_conj2 |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
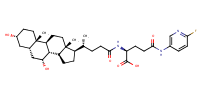
|
glu-CDCA-aminopyridine_conj20 |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
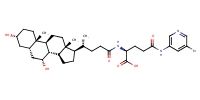
|
glu-CDCA-aminopyridine_conj21 |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
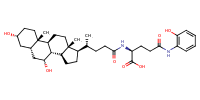
|
glu-CDCA-aminopyridine_conj25 |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
|
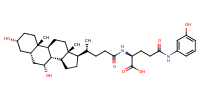
glu-CDCA-aminopyridine_conj26 |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |

|
glu-CDCA-aminopyridine_conj27 |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |

|
glu-CDCA-aminopyridine_conj3 |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
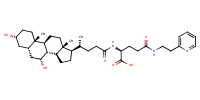
|
glu-CDCA-aminopyridine_conj4 |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |

|
glu-CDCA-aminopyridine_conj5 |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
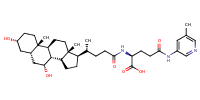
|
glu-CDCA-aminopyridine_conj7 |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |

|
glu-CDCA-aminopyridine_conj8 |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |

|
glu-CDCA-aminopyridine_conj9 |
substrate
|
PMID: 23269503
10.1007/s11095-012-0935-x
AbstractPURPOSE: Membrane transporters mediate many biological effects of chemicals and play a major role in pharmacokinetics and drug resistance. The selection of viable drug candidates among biologically active compounds requires the assessment of their transporter interaction profiles. METHODS: Using public sources, we have assembled and curated the largest, to our knowledge, human intestinal transporter database (>5,000 interaction entries for >3,700 molecules). This data was used to develop thoroughly validated classification Quantitative Structure-Activity Relationship (QSAR) models of transport and/or inhibition of several major transporters including MDR1, BCRP, MRP1-4, PEPT1, ASBT, OATP2B1, OCT1, and MCT1. RESULTS: QSAR models have been developed with advanced machine learning techniques such as Support Vector Machines, Random Forest, and k Nearest Neighbors using Dragon and MOE chemical descriptors. These models afforded high external prediction accuracies of 71-100% estimated by 5-fold external validation, and showed hit retrieval rates with up to 20-fold enrichment in the virtual screening of DrugBank compounds. CONCLUSIONS: The compendium of predictive QSAR models developed in this study can be used for virtual profiling of drug candidates and/or environmental agents with the optimal transporter profiles.
|
external dataset |
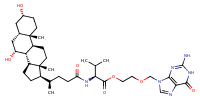
|
Acyclovir valylchenodeoxycholic acid |
substrate
Uptake = 12.9 pmol min-1 (mg of protein)-1
COS
|
PMID: 15832499
10.1021/mp034010t
AbstractThe objective of this work was to design an acyclovir prodrug that would utilize the human apical sodium-dependent bile acid transporter (hASBT) and enhance acyclovir oral bioavailability. Using each chenodeoxycholate, deoxycholate, cholate, and ursodeoxycholate, four bile acid prodrugs of acyclovir were synthesized, where acyclovir was conjugated to a bile acid via a valine linker. The affinity of the prodrug for hASBT was determined through inhibition of taurocholate uptake by COS-7 cells transfected with hASBT (hASBT-COS). The prodrug with the highest inhibitory affinity was further evaluated in vitro and in vivo. The prodrug acyclovir valylchenodeoxycholate yielded the highest affinity for hASBT (Ki = 35 microM), showing that chenodeoxycholate is the free bile acid with the greatest affinity for hASBT. Acyclovir valylchenodeoxycholate's affinity was similar to that of cholic acid (Ki = 25 microM). Further characterization showed that acyclovir was catalytically liberated from acyclovir valylchenode-oxycholate by esterase. Relative to cellular uptake studies of acyclovir alone, the cellular uptake from the prodrug resulted in a 16-fold greater acyclovir accumulation within hASBT-COS cells, indicating enhanced permeation properties of the prodrug. Enhanced permeability was due to hASBT-mediated uptake and increased passive permeability. The extent of acyclovir uptake in the presence of sodium was 1.4-fold greater than the extent of passive prodrug uptake in the absence of sodium (p = 0.02), indicating translocation of the prodrug by hASBT. The prodrug also exhibited an almost 12-fold enhanced passive permeability, relative to acyclovir's passive permeability. Oral administration of acyclovir valylchenodeoxycholate to rats resulted in a 2-fold increase in the bioavailability of acyclovir, compared to the bioavailability after administration of acyclovir alone. Results indicate that a bile acid prodrug strategy may be useful in improving the oral bioavailability of intestinal permeability-limited compounds.
|
literature |